
【中国发明,中国发明授权】一种基于马来酸酐的三元醇超支化单体及其制备方法
有权-审定授权 中国
- 申请号:
- CN201510615692.5
- 专利权人:
- 常州大学
- 授权公告日/公开日:
- 2017.05.10
- 专利有效期:
- 2015.09.24-2035.09.24
- 技术分类:
- C07:有机化学〔2〕
- 转化方式:
- 转让
- 价值度指数:
-
- 63.0分
- 价格:
- 面议
发布人
常州大学
联系人何老师
-

- 0519-88238869

-

- 302910554
-

- 13151263266

- 专利信息&法律状态
- 专利自评
- 专利技术文档
- 价值度指数
- 发明人阵容
 著录项
著录项
- 申请号
- CN201510615692.5
- 申请日
- 20150924
- 公开/公告号
- CN105348091A
- 公开/公告日
- 20160224
- 申请/专利权人
- [常州大学]
- 发明/设计人
- [王克敏, 凌俊杰, 俞强, 高明, 张楠]
- 主分类号
- C07C67/08
- IPC分类号
- C12N 9/0008(2013.01) C12N 9/16
- CPC分类号
- C12N 9/0008(2013.01) C12N 9/16(2013.01)
- 分案申请地址
- 国省代码
- 江苏(32)
- 颁证日
- G06T1/00
- 代理人
摘要
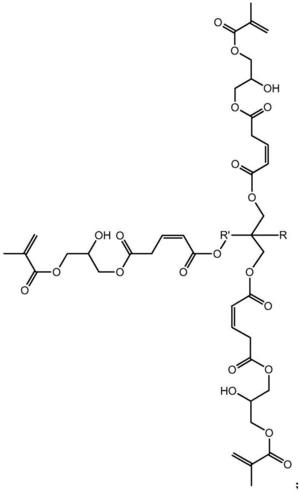
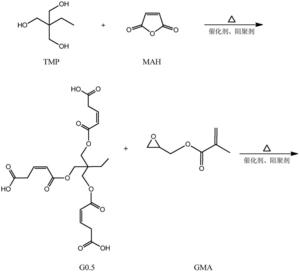
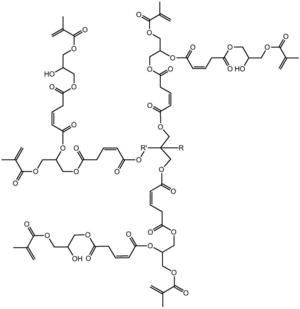
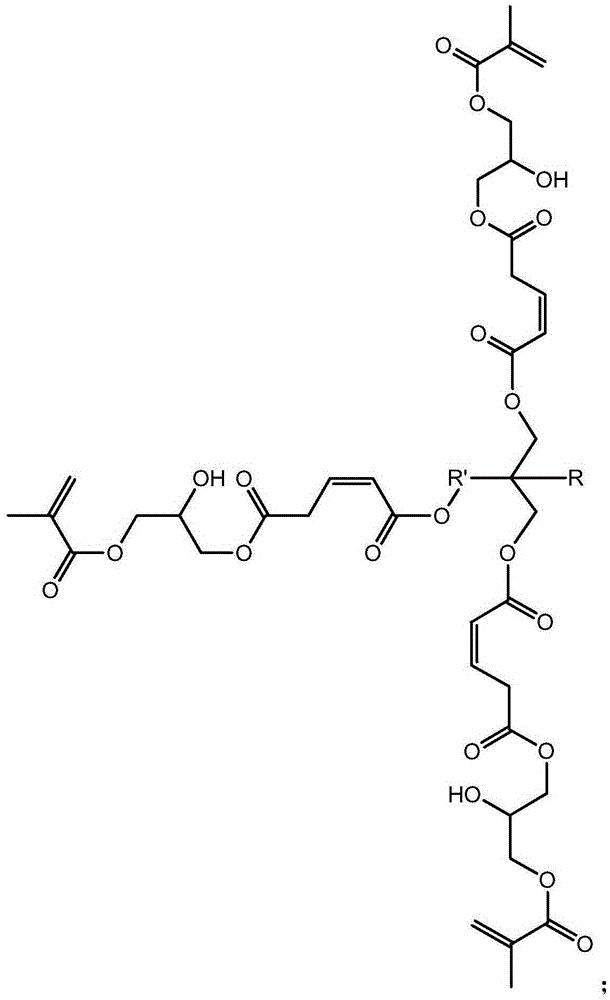
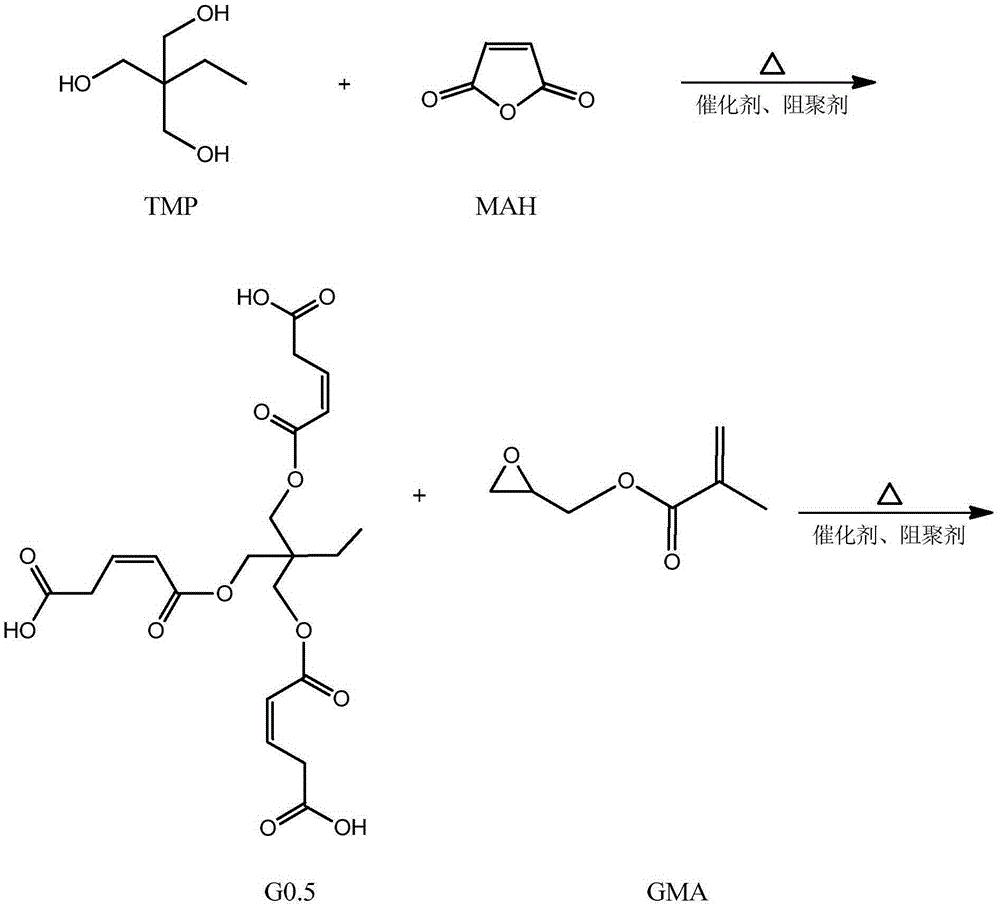
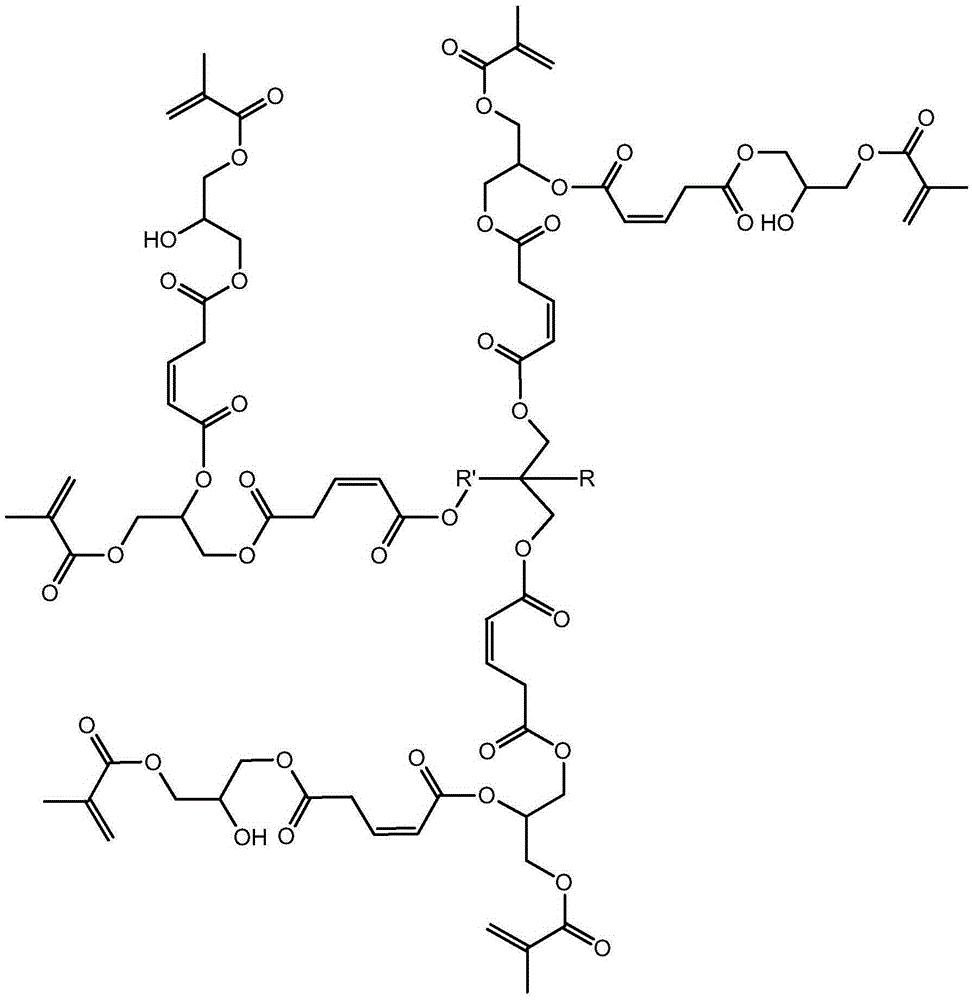
本发明涉及一种基于马来酸酐的三元醇超支化单体,其结构式如下:其中为三元醇。具有(1)该单体是六官能度的,双键转换率高,热稳定好;(2)该单体的附着力较好,硬度较佳,可用于涂料领域,以及其他优点;本发明涉及一种基于马来酸酐的三元醇超支化单体的制备方法,具有反应步骤少,制备周期短,操作简便,易于控制等优点。
法律状态
| 法律状态公告日 | 20240611 |
| 法律状态 | 专利申请权、专利权的转移 |
| 法律状态信息 | 专利权的转移 IPC(主分类):C07C 67/08 专利号:ZL2015106156925 登记生效日:20240527 变更事项:专利权人 变更前权利人:常州校果信息服务有限公司 变更后权利人:南通英韦尔新材料科技有限公司 变更事项:国家或地区 变更前权利人:中国 变更后权利人:中国 变更事项:地址 变更前权利人:213100 江苏省常州市武进高新技术产业开发区常武南路588号天安数码城C幢407-2-6室 变更后权利人:226500 江苏省南通市如皋市磨头镇邓高路19号 |
| 法律状态公告日 | 20211231 |
| 法律状态 | 专利申请权、专利权的转移 |
| 法律状态信息 | 专利权的转移 IPC(主分类):C07C 67/08 专利号:ZL2015106156925 登记生效日:20211217 变更事项:专利权人 变更前权利人:常州大学 变更后权利人:常州校果信息服务有限公司 变更事项:地址 变更前权利人:213164 江苏省常州市武进区滆湖路1号 变更后权利人:213100 江苏省常州市武进高新技术产业开发区常武南路588号天安数码城C幢407-2-6室 |
| 法律状态公告日 | 20170510 |
| 法律状态 | 授权 |
| 法律状态信息 | 授权 |
| 法律状态公告日 | 20160323 |
| 法律状态 | 实质审查的生效 |
| 法律状态信息 | 实质审查的生效 IPC(主分类):C07C 67/08 申请日:20150924 |
| 法律状态公告日 | 20160224 |
| 法律状态 | 公开 |
| 法律状态信息 | 公开 |
| 事务数据公告日 | 20240611 |
| 事务数据类型 | 专利申请权、专利权的转移 |
| 转让详情 | 专利权的转移 IPC(主分类):C07C 67/08 专利号:ZL2015106156925 登记生效日:20240527 变更事项:专利权人 变更前权利人:常州校果信息服务有限公司 变更后权利人:南通英韦尔新材料科技有限公司 变更事项:国家或地区 变更前权利人:中国 变更后权利人:中国 变更事项:地址 变更前权利人:213100 江苏省常州市武进高新技术产业开发区常武南路588号天安数码城C幢407-2-6室 变更后权利人:226500 江苏省南通市如皋市磨头镇邓高路19号 |
| 事务数据公告日 | 20211231 |
| 事务数据类型 | 专利申请权、专利权的转移 |
| 转让详情 | 专利权的转移 IPC(主分类):C07C 67/08 专利号:ZL2015106156925 登记生效日:20211217 变更事项:专利权人 变更前权利人:常州大学 变更后权利人:常州校果信息服务有限公司 变更事项:地址 变更前权利人:213164 江苏省常州市武进区滆湖路1号 变更后权利人:213100 江苏省常州市武进高新技术产业开发区常武南路588号天安数码城C幢407-2-6室 |
权利要求
权利要求数量(8)
独立权利要求数量(2)
1.一种基于马来酸酐的三元醇超支化单体,其结构式如下:
其中 为三元醇,其中R是C nH 2n+1,n≥0,R'是C nH 2n,n≥ 1。
2.如权利要求1所述的一种基于马来酸酐的三元醇超支化单体,其特征在 于:所述三元醇为丙三醇、三羟甲基丙烷、聚醚三元醇中的一种。
3.一种如权利要求1或2所述的基于马来酸酐的三元醇超支化单体的制备 方法,包括如下步骤:
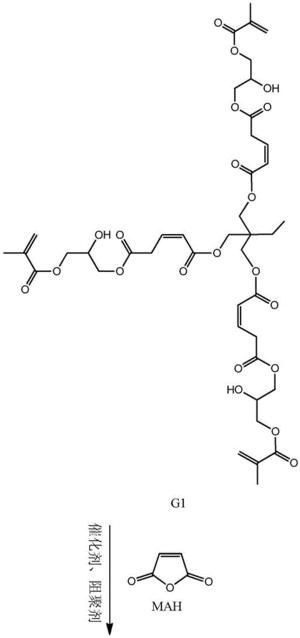
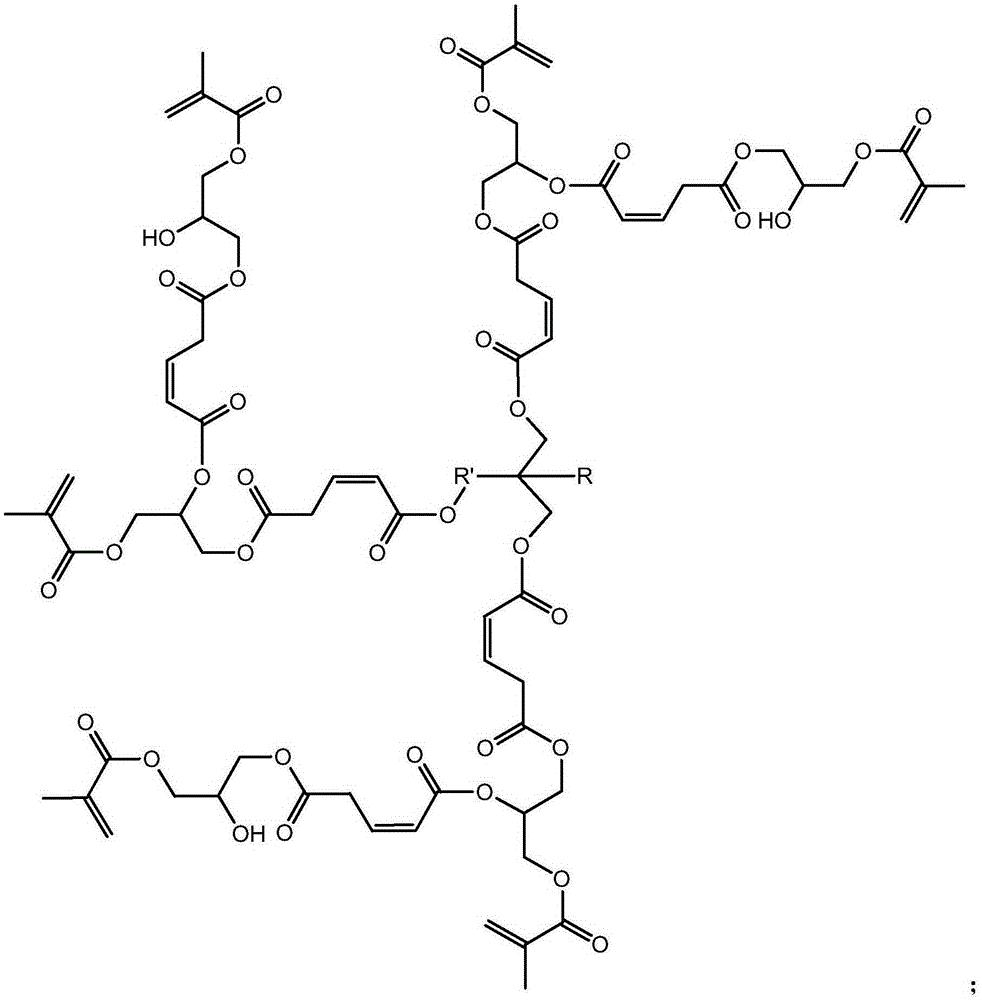
步骤一:以三元醇、马来酸酐和甲基丙烯酸甘油酯为原料制备第一代产物, 其结构式为:
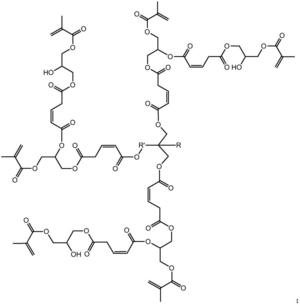
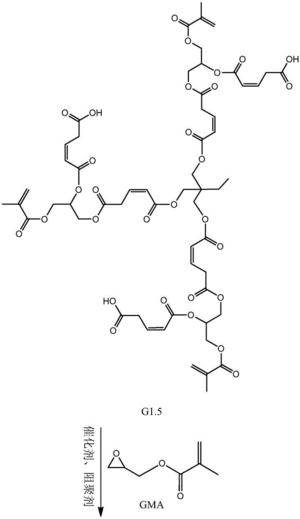
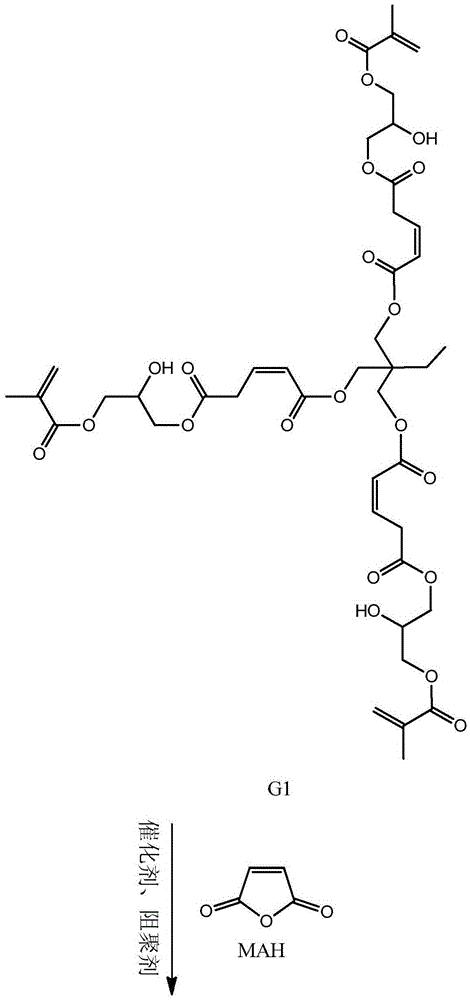
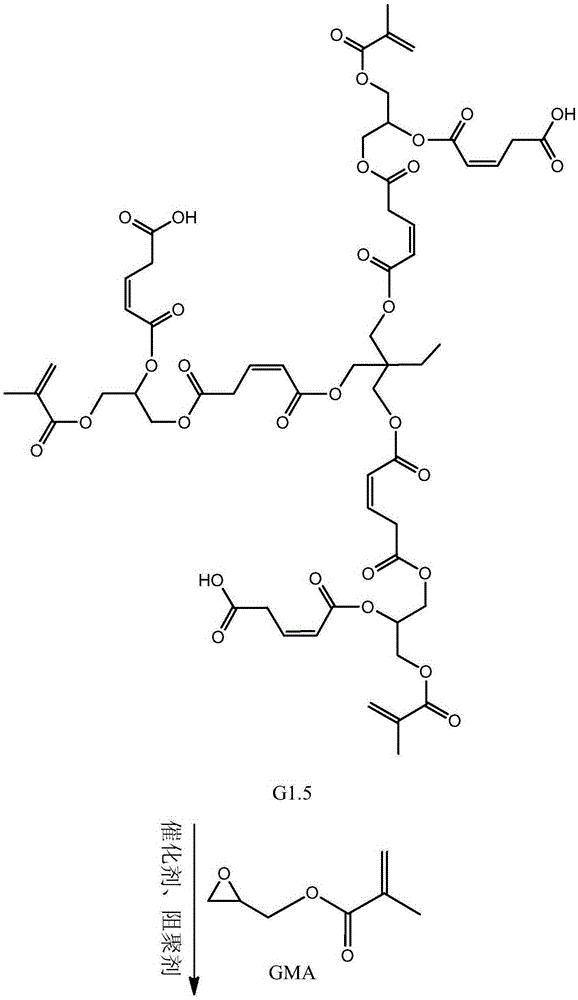
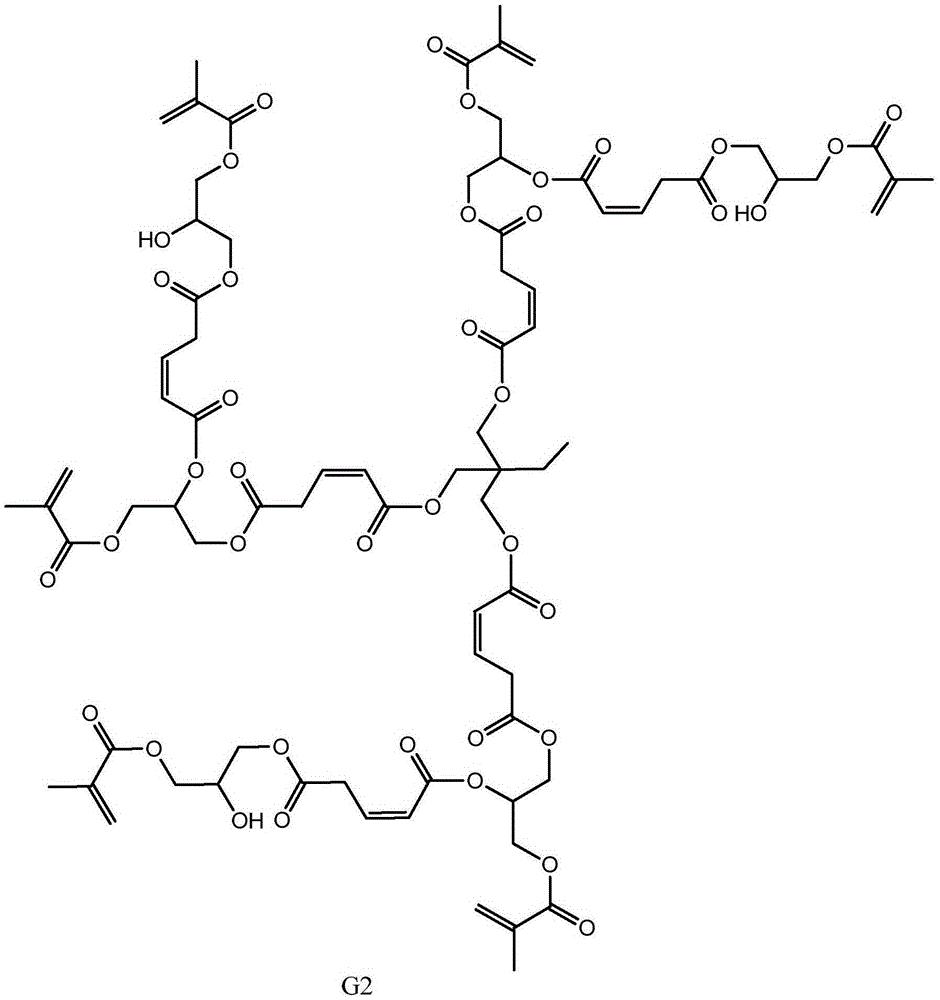
步骤二:以第一代产物、马来酸酐和甲基丙烯酸甘油酯为原料制备第二代 产物,其结构式为:
4.如权利要求3所述的一种基于马来酸酐的三元醇超支化单体的制备方法, 其特征在于:所述步骤一的具体步骤为:
(1)将1份三元醇,3-4份马来酸酐加入到反应容器中,混合均匀,升温 至70-75℃,并加入催化剂和阻聚剂,待马来酸酐融化后,升温至80-85℃,继 续搅拌5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤, 干燥,去除溶剂,得到中间产物;
(2)将3-4份甲基丙烯酸缩水甘油酯,催化剂和溶剂加入到反应容器中, 升温至70℃,将1份步骤(1)中的中间产物逐滴滴入反应器中,在0.5-1h内滴 完,然后将反应体系升温至85℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,水洗2~3次,过滤,干燥,去除溶剂,得到第一代产物。
6.如权利要求4-5任一项所述的一种基于马来酸酐的三元醇超支化单体的 制备方法,其特征在于:所述步骤(1)中的催化剂为浓硫酸、浓盐酸、对甲苯 磺酸、甲烷磺酸和氯化亚砜中的一种,其加入的重量为反应体系的2-5wt%。
7.如权利要求4-5任一项所述的一种基于马来酸酐的三元醇超支化单体的 制备方法,其特征在于:所述步骤(1)阻聚剂为对羟基苯甲醚、氯化亚铜、对 苯二酚、对苯醌和2,5-二甲基对苯二酚中的一种,其加入的重量为反应体系的 1-5wt‰。
8.如权利要求4-5任一项所述的一种基于马来酸酐的三元醇超支化单体的 制备方法,其特征在于:所述步骤(2)中的催化剂为四丁基溴化铵、四甲基氯 化铵、三乙胺、N,N-二甲基苯胺和三苯基磷中的一种,其加入的重量为反应体 系的2-5wt%。
5.如权利要求3所述的一种基于马来酸酐的三元醇超支化单体的制备方法, 其特征在于:所述步骤二的具体步骤为:
(1)将1份第一代产物,3-4份马来酸酐加入到反应容器中,混合均匀, 升温至70-75℃,并加入催化剂和阻聚剂,马来酸酐融化后,升温至80-85℃, 继续搅拌5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤, 干燥,去除溶剂,得到中间产物;
(2)将1-2份甲基丙烯酸缩水甘油酯,催化剂和溶剂加入到反应容器中, 升温至70℃,将1份步骤(3)中的中间产物逐滴滴入反应器中,在0.5-1h内滴 完,然后将反应体系升温至85℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,再用水洗2~3次,过滤,干燥,去除溶剂,得到第二代产物。
1.一种基于马来酸酐的三元醇超支化单体,其结构式如下:
其中为三元醇,其中R是CnH2n+1,n≥0,R'是CnH2n,n≥ 1。
2.如权利要求1所述的一种基于马来酸酐的三元醇超支化单体,其特征在 于:所述三元醇为丙三醇、三羟甲基丙烷、聚醚三元醇中的一种。
3.一种如权利要求1或2所述的基于马来酸酐的三元醇超支化单体的制备 方法,包括如下步骤:
步骤一:以三元醇、马来酸酐和甲基丙烯酸甘油酯为原料制备第一代产物, 其结构式为:
步骤二:以第一代产物、马来酸酐和甲基丙烯酸甘油酯为原料制备第二代 产物,其结构式为:
4.如权利要求3所述的一种基于马来酸酐的三元醇超支化单体的制备方法, 其特征在于:所述步骤一的具体步骤为:
(1)将1份三元醇,3-4份马来酸酐加入到反应容器中,混合均匀,升温 至70-75℃,并加入催化剂和阻聚剂,待马来酸酐融化后,升温至80-85℃,继 续搅拌5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤, 干燥,去除溶剂,得到中间产物;
(2)将3-4份甲基丙烯酸缩水甘油酯,催化剂和溶剂加入到反应容器中, 升温至70℃,将1份步骤(1)中的中间产物逐滴滴入反应器中,在0.5-1h内滴 完,然后将反应体系升温至85℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,水洗2~3次,过滤,干燥,去除溶剂,得到第一代产物。
5.如权利要求3所述的一种基于马来酸酐的三元醇超支化单体的制备方法, 其特征在于:所述步骤二的具体步骤为:
(1)将1份第一代产物,3-4份马来酸酐加入到反应容器中,混合均匀, 升温至70-75℃,并加入催化剂和阻聚剂,马来酸酐融化后,升温至80-85℃, 继续搅拌5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤, 干燥,去除溶剂,得到中间产物;
(2)将1-2份甲基丙烯酸缩水甘油酯,催化剂和溶剂加入到反应容器中, 升温至70℃,将1份步骤(3)中的中间产物逐滴滴入反应器中,在0.5-1h内滴 完,然后将反应体系升温至85℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,再用水洗2~3次,过滤,干燥,去除溶剂,得到第二代产物。
6.如权利要求4-5任一项所述的一种基于马来酸酐的三元醇超支化单体的 制备方法,其特征在于:所述步骤(1)中的催化剂为浓硫酸、浓盐酸、对甲苯 磺酸、甲烷磺酸和氯化亚砜中的一种,其加入的重量为反应体系的2-5wt%。
7.如权利要求4-5任一项所述的一种基于马来酸酐的三元醇超支化单体的 制备方法,其特征在于:所述步骤(1)阻聚剂为对羟基苯甲醚、氯化亚铜、对 苯二酚、对苯醌和2,5-二甲基对苯二酚中的一种,其加入的重量为反应体系的 1-5wt‰。
8.如权利要求4-5任一项所述的一种基于马来酸酐的三元醇超支化单体的 制备方法,其特征在于:所述步骤(2)中的催化剂为四丁基溴化铵、四甲基氯 化铵、三乙胺、N,N-二甲基苯胺和三苯基磷中的一种,其加入的重量为反应体 系的2-5wt%。
说明书
技术领域
本发明涉及感光高分子材料技术领域,尤其涉及一种基于马来酸酐的超支 化丙烯酸脂作为光聚合单体。
背景技术
光聚合(又称光固化)技术是利用光(紫外光或可见光)引发具有化学反应活 性的液态物质快速转变为固态物质的过程,是20世纪60年代问世的新型绿色 技术。1946年,美国Inmont公司首次发表了不饱和聚酯/苯乙烯UV固化油墨专 利,到80年代末,UV固化技术一直保持着年均15%以上的增长率。进入20世 纪90年代中期以来,仍以每年接近10%的速度快速增长,在某些领域还有增长 趋势。光固化技术具有高效、适应性广、经济、节能、环境友好等特点,这些 特点符合当今世界各国对环保、节能的要求,广泛应用于涂料、油墨、胶粘剂、 成像、微电子、齿科修复和生物材料等领域。
光固化技术与传统的热固化技术不同之处在于:光固化反应本质是由紫外 光引发的聚合、交联反应,任何一个光固化体系至少包括以下三个部分:(1) 低聚物(或称预聚物、树脂),赋予材料以基本的物理化学性能;(2)单体,又 称活性稀释剂,主要用于调节体系的黏度,但是对固化速率和材料的性能也有 影响;(3)光引发剂,用于产生引发聚合反应的活性种(自由基或阳离子)。
活性稀释剂一般是含有可聚合官能团的小分子,因而在业内习惯上也称之 为“单体”。活性稀释剂通常能参与聚合交联过程,不像传统的溶剂型涂料、 油墨中的有机溶剂那样挥发到空气中,因此,这一优点赋予了光固化体系的环 保特性。活性稀释剂按其每个分子所含反应性基团的多少,可以分为单官能团 活性稀释剂和多官能团活性稀释剂。单官能团活性稀释剂每个分子中仅含一个 可参与固化反应的基团,如甲基丙烯酸—β—羟乙酯(HEMA)。多官能团活性稀 释剂是指每个分子中含有两个或两个以上可参与固化反应基团的活性稀释剂, 如1,6-己二醇二丙烯酸酯(HDDA)。采用含较多官能团的单体,除了增加反应活 性外,还能赋予固化膜交联结构。
在辐射固化组成中,单体起着关键的作用。除了调节体系的粘度以外,它 还能影响到固化动力学,聚合程度以及所生成聚合物的物理机械性质等等。虽 然固化材料的性质基本上由所使用的低聚物决定,但是主要的技术和安全问题 却必须考虑到所用单体的性质。
按固化机理,活性稀释剂可分为自由基型和阳离子型两类。(甲基)丙烯酸 酯类是典型的自由基型活性稀释剂,固化反应通过自由基光聚合进行。环氧类 则属于阳离子型活性稀释剂,其固化反应机理则是阳离子聚合反应。而乙烯基 醚类既可参与自由基聚合,也可进行阳离子聚合,因此可作为两种光固化体系 的活性稀释剂。
在光固化配方体系中,活性稀释剂和低聚物一起占整个配方质量的90%以 上,并且决定成型后材料基本的物理化学性能。理想的单体具有以下特点:(1) 聚合收缩小、固化程度高(即双键转化率高)且不会降低固化后材料的机械性 能;(2)疏水性好;(3)廉价,合成简单;(4)稳定性好,便于长时间保存。
早期使用的单官能度单体中,如丙烯酸正丁酯(n-BA)、丙烯酸异丁酯(i-BA) 均为高挥发性稀释剂,早期主要用于配制木器漆,具有易燃、气味大、固化速 度低等缺陷,因此现在已经很少采用。而双官能度单体含有两个光活性的(甲基) 丙烯酸酯官能团。固化速度快于单官能团单体,成膜的交联密度随交联点的增 加随之增大,但仍保持良好的稀释效果。另外,随单体官能度增加,分子量变 大,其挥发性逐渐减小,气味也降低。DuyguAvci等以EHMA、HEA和HEMA为原 料,与C18双丙烯酰氯反应得到一系列带羟甲基的双官能团(甲基)丙烯酸酯类单 体。同时用DSC对其光聚合行为进行了研究。这类单体含有柔韧的长链结构, 可以增强材料的耐冲击性和韧性,扩宽了其应用领域。 (DuyguA,JenniferN,LonJM.Synthesisandphotopolymerizationkineticsof newflexiblediacrylatecrosslinkersbasedonC18diacid[J].Polymer, 2003,44:963-968.)一般来说丙烯酸酯类单体光固化后收缩率大,耐热性较差, 使光固化材料的应用领域受到影响。因此,设计及开发新型功能性丙烯酸酯类 光活性单体对拓展光固化材料的应用领域及制备高性能的光固化材料具有重要 的意义。传统的氨基甲酸酯(甲基)丙烯酸酯一般都是通过异氰酸酯与带羟基的 化合物进行加成反应制备而得的。但是异氰酸酯有刺激性气味,对皮肤、眼睛和 呼吸道有强烈的刺激作用,毒副作用比较大。
发明内容
本发明的目的在于提供一种基于马来酸酐的三元醇超支化单体,具有以下 几个优点:
(1)该单体是六官能度的,双键转换率高,热稳定好;
(2)该单体的附着力较好,硬度较佳,可用于涂料领域;
本发明的目的在于提供一种基于马来酸酐的三元醇超支化单体的制备方 法,具有以下几个优点:
(1)反应步骤少,制备周期短,操作简便,易于控制。
本发明解决其技术问题所采用的技术方案是:一种基于马来酸酐的三元醇 超支化单体,其结构式如下:
其中为三元醇,其中R是CnH2n+1,n≥0,R'是CnH2n,n≥1。
具体地,所述三元醇为丙三醇、三羟甲基丙烷、聚醚三元醇中的一种。
一种如上所述的基于马来酸酐的三元醇超支化单体的制备方法,包括如下 步骤:
步骤一:以三元醇、马来酸酐和甲基丙烯酸甘油酯为原料制备第一代产物, 其结构式为:
步骤二:以第一代产物、马来酸酐和甲基丙烯酸甘油酯为原料制备第二代 产物,即为目标产物:基于马来酸酐的三元醇超支化单体,其结构式为:
作为优选,所述步骤一的具体步骤为:
(1)将1份三元醇,3-4份马来酸酐加入到反应容器中,混合均匀,升温 至70-75℃,并加入催化剂和阻聚剂,待马来酸酐融化后,升温至80-85℃,继 续搅拌5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤, 干燥,去除溶剂,得到中间产物;
(2)将3-4份甲基丙烯酸缩水甘油酯,催化剂和溶剂加入到反应容器中, 升温至70℃,将1份步骤(1)中的中间产物逐滴滴入反应器中,在0.5h内滴 完,然后将反应体系升温至85℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,水洗2~3次,过滤,干燥,去除溶剂,得到第一代产物。
作为优选,所述步骤二的具体步骤为:
(1)将1份第一代产物,3-4份马来酸酐加入到反应容器中,混合均匀, 升温至70-75℃,并加入催化剂和阻聚剂,马来酸酐融化后,升温至80-85℃, 继续搅拌5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤, 干燥,去除溶剂,得到中间产物;
(2)将3-4份甲基丙烯酸缩水甘油酯,催化剂和溶剂加入到反应容器中, 升温至70℃,将1份步骤(1)中的中间产物逐滴滴入反应器中,在0.5h内滴 完,然后将反应体系升温至85℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,再用水洗2~3次,过滤,干燥,去除溶剂,得到第二代产物。
具体地,所述步骤(1)中的催化剂为浓硫酸、浓盐酸、对甲苯磺酸、甲烷 磺酸和氯化亚砜中的一种,其加入的重量为反应体系的2-5wt%。
具体地,所述步骤(1)阻聚剂为对羟基苯甲醚、氯化亚铜、对苯二酚、对 苯醌和2,5-二甲基对苯二酚中的一种,其加入的重量为反应体系的1-5wt‰。
具体地,所述步骤(2)中的催化剂为四丁基溴化铵、四甲基氯化铵、三乙 胺、N,N-二甲基苯胺和三苯基磷中的一种,其加入的重量为反应体系的2-5wt%。
本发明从分子结构设计出发,(以三羟甲基丙烷为例)首先三羟甲基丙烷上 的三个羟基打开马来酸酐上的酸酐结构,生成中间产物G0.5,然后通过G0.5上 的三个羧基打开GMA上的环氧结构,得到一代产物G1,接着G1上的三个羟基打 开马来酸酐上的酸酐结构,生成中间产物G1.5,然后G1.5上的三个羧基打开 GMA上的环氧结构,得到二代产物,即最终产物G2。
本发明所制备一种基于马来酸酐的三元醇超支化单体的整个反应方程式可 表示为:(以三羟甲基丙烷为例)
附图说明
下面结合附图和实施例对本发明进一步说明。
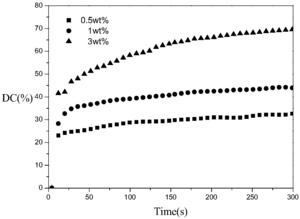
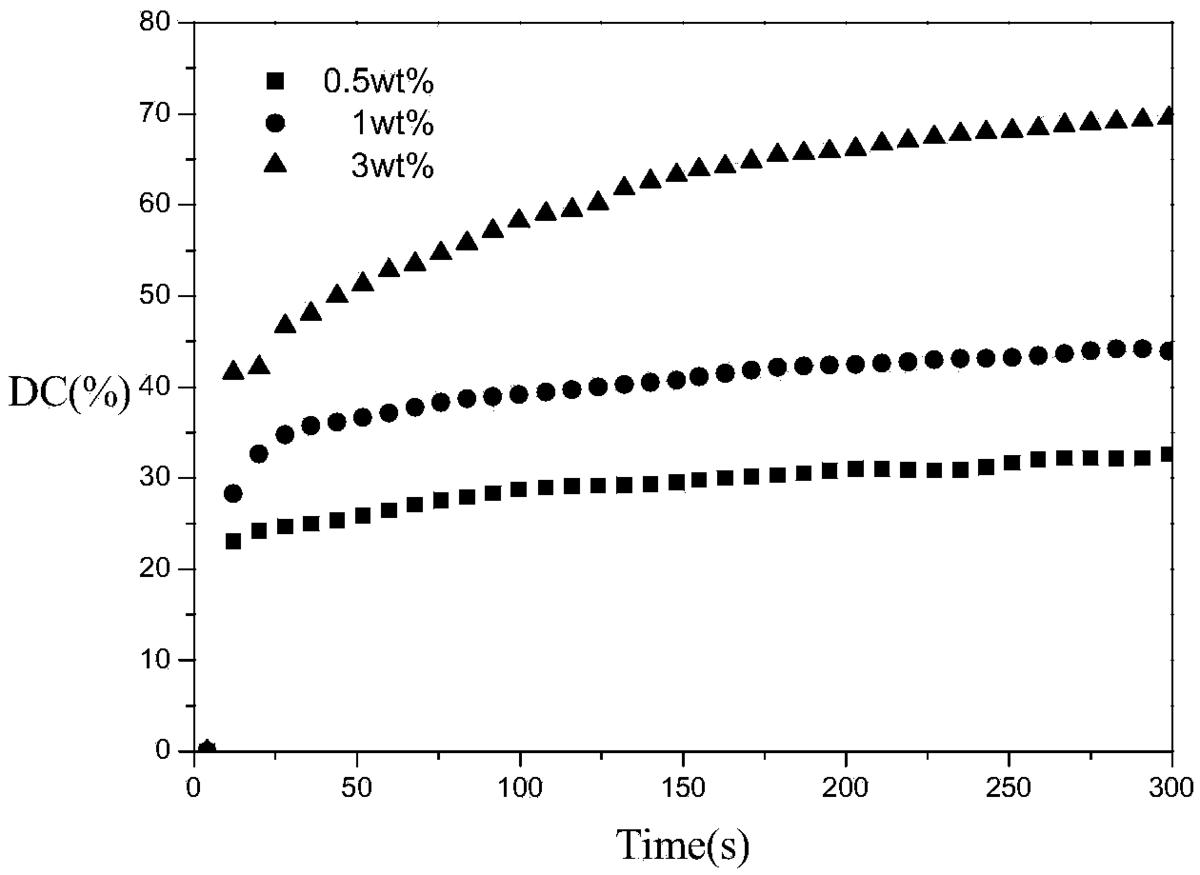
图1是裂解型引发剂184引发实施例1得到的G2的光聚合双键转化率—时 间图;
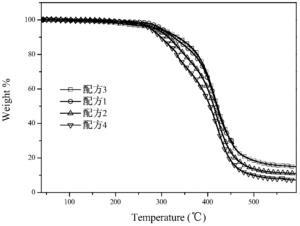
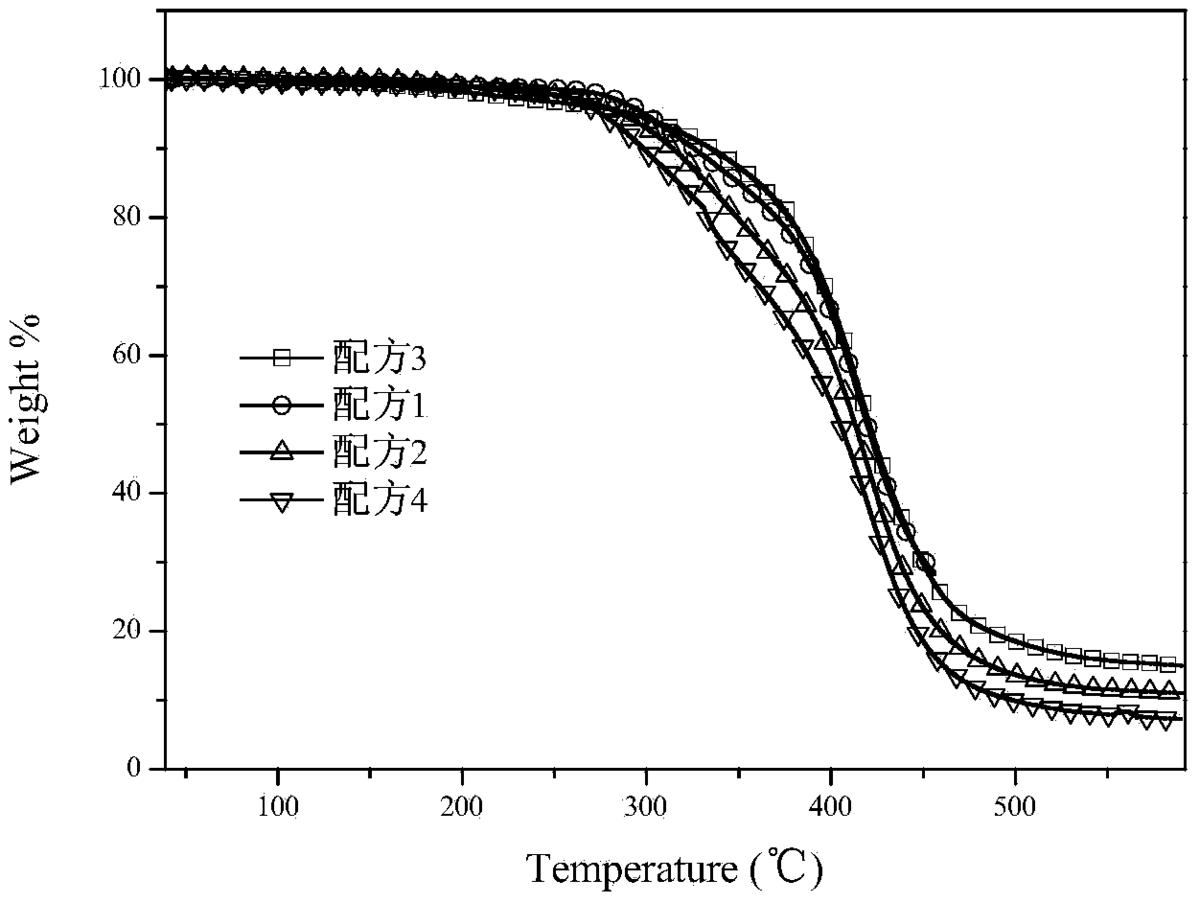
图2是各配方随着温度升高热失重曲线图。
具体实施方式
现在结合附图对本发明作进一步详细的说明。这些附图均为简化的示意图, 仅以示意方式说明本发明的基本结构,因此其仅显示与本发明有关的构成。
实施例1
(1)将0.1mol(13.4g)三羟甲基丙烷,0.3mol(29.4g)马来酸酐加入到 反应容器中,混合均匀,搅拌升温至70℃,并加入催化剂对甲苯磺酸(0.86g, 反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.21g,反应体系的5wt‰),待马 来酸酐融化后,升温至80℃,继续搅拌5h后,用乙酸乙酯萃取产物,再用水洗 2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到一代产物G0.5。
(2)将0.3mol(42.66g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁基 氯化铵(1.61g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.41g,反应体系 的5wt‰)加入到反应容器中,升温至70℃,将1份G0.5(0.1mol,47.05g) 逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至80℃,继续搅拌 5h后,冷却至室温,用乙酸乙酯萃取产物,水洗2~3次,过滤,并用无水硫酸 钠干燥,旋蒸,得到第一代产物G1。
(3)将0.1mol(89.6g)的第一代产物G1,0.3mol(29.4g)的马来酸酐 加入到反应容器中,混合均匀,搅拌升温至60℃,并加入催化剂对甲苯磺酸 (2.38g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.59g,反应体系的5wt‰), 待马来酸酐融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,再用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到 中间产物G1.5。
(4)将0.3mol(14.25g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁基 氯化铵(1.11g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.28g,反应体系 的5wt‰)加入到反应容器中,升温至70℃,将1份G1.5(0.1mol,41.2g)逐 滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至80℃,继续搅拌5h 后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤,无水硫酸钠 干燥,减压蒸馏除去溶剂,得到第二代产物G2。即目标产物55.44g。产品基本 物理性质测试:黏度(cps25℃)520,色度(APHA/Gardner)178,折射率1.217, 表面张力(Dynes/cm20℃)52.3。
实施例2
(1)将0.1mol(13.4g)三羟甲基丙烷,0.3mol(29.4g)马来酸酐加入到 反应容器中,混合均匀,搅拌升温至60℃,并加入催化剂对甲苯磺酸(0.86g, 反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.21g,反应体系的5wt‰),待马 来酸酐融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃取产 物,再用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到中间 产物G0.5。
(2)将0.31mol(44.08g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁 基氯化铵(1.82g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.46g,反应体 系的5wt‰)加入到反应容器中,升温至70℃,将1份步骤(1)中的反应产物 (0.1mol,47.05g)逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温 至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3 次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到第一代产物G1。
(3)将0.1mol(79.6g)的第一代产物G1,0.3mol(26.09g)的马来酸酐 加入到反应容器中,混合均匀,搅拌升温至60℃,并加入催化剂对甲苯磺酸 (2.11g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.53g,反应体系的5wt‰), 待马来酸酐融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,再用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到 中间产物G1.5。
(4)将0.31mol(13.91g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁 基氯化铵(1.08g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.27g,反应体 系的5wt‰)加入到反应容器中,升温至70℃,将1份G1.5(0.1mol,40.2g) 逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至80℃,继续搅拌 5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤,无水硫酸 钠干燥,减压蒸馏除去溶剂,得到第二代产物G2。即目标产物54.09g。产品基 本物理性质测试:黏度(cps25℃)515,色度(APHA/Gardner)177,折射率 1.2248,表面张力(Dynes/cm20℃)55.2。
实施例3
(1)将0.1mol(13.4g)三羟甲基丙烷,0.3mol(29.4g)马来酸酐加入到 反应容器中,混合均匀,搅拌升温至60℃,并加入催化剂对甲苯磺酸(0.86g, 反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.21g,反应体系的5wt‰),待马 来酸酐融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃取产 物,再用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到中间 产物G0.5。
(2)将0.32mol(45.51g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁 基氯化铵(1.85g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.46g,反应体 系的5wt‰)加入到反应容器中,升温至70℃,将1份G0.5(0.1mol,47.05g) 逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至80℃,继续搅拌 5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤,无水硫酸 钠干燥,减压蒸馏除去溶剂,得到第一代产物G1。
(3)将0.1mol(78.4g)的第一代产物G1,0.3mol(25.69g)的马来酸酐 加入到反应容器中,混合均匀,搅拌升温至60℃,并加入催化剂对甲苯磺酸 (2.08g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.52g,反应体系的5wt‰), 待马来酸酐融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,再用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到 中间产物G1.5。
(4)将0.32mol(13.67g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁 基氯化铵(1.09g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.27g,反应体 系的5wt‰)加入到反应容器中,升温至70℃,将1份G1.5(0.1mol,39.5g) 逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至80℃,继续搅拌 5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤,无水硫酸 钠干燥,减压蒸馏除去溶剂,得到第二代产物G2。即目标产物53.16g。产品基 本物理性质测试:黏度(cps25℃)510,色度(APHA/Gardner)174,折射率 1.2697,表面张力(Dynes/cm20℃)54.9。
实施例4
(1)将0.1mol(13.4g)三羟甲基丙烷,0.31mol(30.38g)马来酸酐加入 到反应容器中,混合均匀,搅拌升温至70℃,并加入催化剂对甲苯磺酸(0.86g, 反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.21g,反应体系的5wt‰),待马 来酸酐融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃取产 物,再用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到中间 产物G0.5。
(2)将0.32mol(47.77g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁 基氯化铵(1.88g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.47g,反应体 系的5wt‰)加入到反应容器中,升温至70℃,将1份G0.5(0.1mol,46.25g) 逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至80℃,继续搅拌 5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤,无水硫酸 钠干燥,减压蒸馏除去溶剂,得到第一代产物G1。
(3)将0.1mol(76.4g)的第一代产物G1,0.3mol(25.2g)的马来酸酐 加入到反应容器中,混合均匀,搅拌升温至70℃,并加入催化剂对甲苯磺酸 (2.03g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.51g,反应体系的5wt‰), 待马来酸酐融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,再用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到 中间产物G1.5。
(4)将0.32mol(15.61g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁 基氯化铵(1.16g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.29g,反应体 系的5wt‰)加入到反应容器中,升温至70℃,将1份G1.5(0.1mol,42.3g) 逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至80℃,继续搅拌 5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤,无水硫酸 钠干燥,减压蒸馏除去溶剂,得到第二代产物G2。即目标产物56.93g。产品基 本物理性质测试:黏度(cps25℃)505,色度(APHA/Gardner)171,折射率 1.2469,表面张力(Dynes/cm20℃)56.4。
实施例5
(1)将0.1mol(13.4g)正丁醇,0.33mol(32.34g)马来酸酐加入到反应 容器中,混合均匀,搅拌升温至70℃,并加入催化剂对甲苯磺酸(0.91g,反应 体系的3wt%)和阻聚剂对羟基苯甲醚(0.18g,反应体系的4wt‰),待马来酸 酐融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃取产物, 再用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到中间产物 G0.5。
(2)将0.12mol(42.67g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁 基氯化铵(1.74g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.43g,反应体 系的5wt‰)加入到反应容器中,升温至70℃,将1份G0.5(0.1mol,44.13g) 逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至85℃,继续搅拌 5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤,无水硫酸 钠干燥,减压蒸馏除去溶剂,得到第一代产物G1。
(3)将0.1mol(73.3g)的第一代产物G1,0.33mol(26.43g)的马来酸 酐加入到反应容器中,混合均匀,搅拌升温至70℃,并加入催化剂对甲苯磺酸 (2.99g,反应体系的3wt%)和阻聚剂对羟基苯甲醚(0.39g,反应体系的4wt‰), 待马来酸酐融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,再用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂G1.5。
(4)将0.32mol(16.27g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁 基氯化铵(1.21g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.31g,反应体 系的5wt‰)加入到反应容器中,升温至70℃,将1份G1.5(0.1mol,44.1g) 逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至85℃,继续搅拌 5h后,冷却至室温用乙酸乙酯萃取产物,再用水洗2~3次,过滤,无水硫酸钠 干燥,减压蒸馏除去溶剂,得到第二代产物G2。即目标产物59.35g。产品基本 物理性质测试:黏度(cps25℃)500,色度(APHA/Gardner)170,折射率1.2897, 表面张力(Dynes/cm20℃)50.3。
实施例6
(1)将0.1mol(13.4g)正丁醇,0.35mol(34.3g)马来酸酐加入到反应 容器中,混合均匀,搅拌升温至70℃,并加入催化剂对甲苯磺酸(1.91g,反应 体系的4wt%)和阻聚剂对羟基苯甲醚(2.38g,反应体系的5wt‰),待马来酸 酐融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃取产物, 再用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到中间产物 G0.5。
(2)将0.35mol(49.96g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁 基氯化铵(1.94g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.49g,反应体 系的5wt‰)加入到反应容器中,升温至70℃,将1份G0.5(0.1mol,46.23g) 逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至85℃,继续搅拌 5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤,无水硫酸 钠干燥,减压蒸馏除去溶剂,得到第一代产物G1。
(3)将0.1mol(71.3g)的第一代产物G1,0.35mol(27.3g)的马来酸酐 加入到反应容器中,混合均匀,搅拌升温至70℃,并加入催化剂对甲苯磺酸 (3.94g,反应体系的4wt%)和阻聚剂对羟基苯甲醚(4.93g,反应体系的5wt‰), 待马来酸酐融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃 取产物,再用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到 中间产物G1.5。
(4)将0.35mol(18.65g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁 基氯化铵(1.29g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(3.24g,反应体 系的5wt‰)加入到反应容器中,升温至70℃,将1份G1.5(0.1mol,46.2g) 逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至85℃,继续搅拌 5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3次,过滤,无水硫酸 钠干燥,减压蒸馏除去溶剂,得到第二代产物G2。即目标产物62.18g。产品基 本物理性质测试:黏度(cps25℃)502,色度(APHA/Gardner)175,折射率 1.3102,表面张力(Dynes/cm20℃)51.3。
实施例7
(1)将0.1mol(13.4g)正丁醇,0.4mol(39.2g)马来酸酐加入到反应容 器中,混合均匀,搅拌升温至70℃,并加入催化剂对甲苯磺酸(1.05g,反应体 系的2wt%)和阻聚剂对羟基苯甲醚(0.26g,反应体系的5wt‰),待马来酸酐 融化后,升温至80℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃取产物,再 用水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到中间产物G0.5。
(2)将0.4mol(48.64g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁基 氯化铵(1.78g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.44g,反应体系 的5wt‰)加入到反应容器中,升温至70℃,将1份G0.5(0.1mol,40.23g) 逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温至85℃,继续搅拌 5h后,冷却至室温,用乙酸乙酯萃取产物,水洗2~3次,过滤,并用无水硫酸 钠干燥,旋蒸,得到第一代产物G1。
(3)将0.1mol(70.7g)的第一代产物G1,0.4mol(39.2g)的马来酸酐 加入到反应容器中,混合均匀,搅拌升温至70℃,并加入催化剂对甲苯磺酸 (2.19g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.33g,反应体系的3wt‰), 待马来酸酐融化后,升温至80℃,继续搅拌5h后,用乙酸乙酯萃取产物,再用 水洗2~3次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到中间产物G1.5。
(4)将0.4mol(18.89g)的甲基丙烯酸缩水甘油酯(GMA),催化剂四丁基 氯化铵(1.19g,反应体系的2wt%)和阻聚剂对羟基苯甲醚(0.29g,反应体系 的5wt‰)加入到反应容器中,升温至70℃,将1份步骤(3)中的反应产物 (0.1mol,40.95g)逐滴滴入反应器中,在0.5h内滴完,然后将反应体系升温 至85℃,继续搅拌5h后,冷却至室温,用乙酸乙酯萃取产物,再用水洗2~3 次,过滤,无水硫酸钠干燥,减压蒸馏除去溶剂,得到第二代产物G2。即目标 产物55.11g。产品基本物理性质测试:黏度(cps25℃)518,色度(APHA/Gardner) 179,折射率1.3508,表面张力(Dynes/cm20℃)54.8。
应用实施例:
固化速率:
以实施例1得到的第二代超支化单体,将三羟甲基丙烷第二代超支化单体 G2,在不同浓度的裂解型光引发剂184配制成聚合体系(引发剂浓度:0.5%、 1%、3%),超声震荡2分钟保证体系混合均匀,然后,取微量样品涂抹在溴化钾 压片上,在室温下用带400-500nm波段滤波片的EFOSLite点光源以500mW/cm2的光强照射样品5分钟,配有水平样品台的RTIR通过监测800-850cm-1丙烯酸酯 双键特征吸收峰峰面积的变化直观地反映聚合体系的程度。其具体配方如下所 示:
配方 二代超支化单体G2 引发剂184 1 0.2g 0.5% 2 0.2g 1% 3 0.2g 3%
其结果如图1所示,从图1中可以看出,G2用紫外灯光照300秒即可完全 聚合,随着引发剂浓度升高,丙烯酸酯双键转化率升高;当引发剂浓度3%时, 丙烯酸酯双键转化率接近70%。
热力学性能:
本发明对G2制备出的配方体系,进行热失重分析,采用一般的牙科树脂配 方体系(Bis-GMA,70%;稀释剂TEGDMA30%;引发剂樟脑坤CQ,1.5wt%;无机填料 SiO2,80%),改变其中的单体(TEGDMA)和G2的组成部分及含量,观察体系的热 稳定性。其结果如图2所示,当G2替代单体TEGDMA时,整个体系的热稳定性 较好,大约在330℃开始分解。
配方 Bis-GMA G2 TEGDMA 2]]> 引发剂184(4%) 1 7g 2g 1g 8g 0.4g 2 7g 1g 2g 8g 0.4g 3 7g 3g 0 8g 0.4g 4 7g 0 3g 8g 0.4g
G2附着力性能:
不同材质附着力测试:(ISO标准-T9999358)
试样材质 等级 PP 4 铁片 0 100mmPET 0 200mmPET 0 50mmPET 0 马口铁 2 普通玻璃 1
G2铅笔硬度测试:
铅笔规格 测试结果
6H 破 5H 破 4H 不破 3H 不破 2H 不破 H 不破 B 不破 HB 不破
以上述依据本发明的理想实施例为启示,通过上述的说明内容,相关工作 人员完全可以在不偏离本项发明技术思想的范围内,进行多样的变更以及修改。 本项发明的技术性范围并不局限于说明书上的内容,必须要根据权利要求范围 来确定其技术性范围。
价值度评估
技术价值
经济价值
法律价值
0 0 063.0分
0 50 75 100专利价值度是通过科学的评估模
型对专利价值进行量化的结果,
基于专利大数据针对专利总体特
征指标利用计算机自动化技术对
待评估专利进行高效、智能化的
分析,从技术、经济和法律价值
三个层面构建专利价值评估体
系,可以有效提升专利价值评估
的质量和效率。
总评:63.0分
该专利价值中等 (仅供参考)
本专利文献中包含【7 个实施例】、【2 个技术分类】,从一定程度上而言上述指标的数值越大可以反映出所述专利的技术保护及应用范围越广。 【专利权的维持时间11 年】专利权的维持时间越长,其价值对于权利人而言越高。 尤其重要是,该专利 【权利转移2 次】、 都从侧面反应出该专利的技术、经济和法律价值。
技术价值 32.0
该指标主要从专利申请的著录信息、法律事件等内容中挖掘其技术价值,专利类型、独立权利要求数量、无效请求次数等内容均可反映出专利的技术性价值。 技术创新是专利申请的核心,若您需要进行技术借鉴或寻找可合作的项目,推荐您重点关注该指标。
部分指标包括:
授权周期(发明)
19 个月独立权利要求数量
2 个从属权利要求数量
5 个说明书页数
15 页实施例个数
7 个发明人数量
5 个被引用次数
0 次引用文献数量
0 个优先权个数
0 个技术分类数量
2 个无效请求次数
0 个分案子案个数
0 个同族专利数
0 个专利获奖情况
无保密专利的解密
否经济价值 9.0
该指标主要指示了专利技术在商品化、产业化及市场化过程中可能带来的预期利益。 专利技术只有转化成生产力才能体现其经济价值,专利技术的许可、转让、质押次数等指标均是其经济价值的表征。 因此,若您希望找到行业内的运用广泛的热点专利技术及侵权诉讼中的涉案专利,推荐您重点关注该指标。
部分指标包括:
申请人数量
1申请人类型
院校许可备案
0 次权利质押
0 次权利转移
2 个海关备案
否法律价值 22.0
该指标主要从专利权的稳定性角度评议其价值。专利权是一种垄断权,但其在法律保护的期间和范围内才有效。 专利权的存续时间、当前的法律状态可反映出其法律价值。故而,若您准备找寻权属稳定且专利权人非常重视的专利技术,推荐您关注该指标。
部分指标包括:
存活期/维持时间
11法律状态
有权-审定授权


 苏公网安备 32041202001399号
苏公网安备 32041202001399号



 loading...
loading...